
Hype vs Reality: The Microbiome
With so much hype today about one’s gut health, you often wonder how much of it is truth. A trip down to your local pharmacy’s supplements shelf and you will see a wide range of pro- and pre- biotics each with their own beneficial claims and often too ludicrous to be taken seriously.
In case you still don’t believe me, just head down to subreddit: r/microbiome with close to 3000 subscribers.
Why this hype? It’s driven by the numerous use cases which are popping up all over the place involving the microbiome.
From prediction for early diagnostics of chronic diseases such as diabetes to finger printing criminals from samples left behind at crime scenes.
So How do we study microbes (even those we cannot culture)?
Through DNA sequencing of course!

More data for you, me, everybody
We study them mainly using two techniques
- Amplicon Sequencing
- Whole Metagenome Sequencing
So What is Amplicon sequencing and Metagenomics?
| The investigation of microbes in a given sample without the need for culture by directly recording the genetic content using next generation DNA sequencing techniques. |
Can you be more specific? (TL;DR version)
- Sequencing of a only a selected representative gene of interest and in this case the variable regions in the rRNA of the 16S ribosomes
- Whole metagenome sequencing, you basically get the whole repertoire / complement of genes.
for non-biologist: You can compare this with many existing data science techniques where you either churn through all collected data or just zoom in onto a very specific signal which you’re looking for
5 things you should know about your gut microbiome
1. Community complexity
Microbial communities range widely in their complexity. By complexity we mean to say the numbers of unique OTUs (Operational Taxanomic Unit) and their proportions.
There’s several ways to quantify this, the complexity, is through unweighted and weighted indices borrowed from existing macroecology literature:
Indices and metrics

There’s many of these around and the famous ones are $\alpha$-diversity and $\beta$- diversity.
The former, $\alpha$-diveristy, measures within sample diversity and includes: Shannon-Weaver, Simpson If you want something more weighted then try Taxonomic Diversity $\Delta$ or Taxonomic Distinctiveness $\Delta^+$. (See the R package: Vegan for more explanation)
$\beta$-diveristy describes the total species diversity across samples over the average species diversity per sample its used essentially as a measure to investigate heterogenity within the data amongst samples.
2. Analysis is Hard
Simpler communities are by far easier to study. However, one must also consider the number of reference genomes available for the community in question, ie. if community was simple but the reference genomes are sparse and few inbetween, chances are the analysis will only unveil little about the community save for top level analyses.
This places the gut microbiome in a good location to be studied as it is relatively well understood with good reference genomes and isn’t as complicated as the soil or wastewater microbiome.

The gut microbiome is right smack in the goldilock zone for analysis
3. Types of Communities
Simple Communities

These are found usually in very harsh or low nutrient environments.
Complex communities

Examples of complex communities include soil, sewage where species numbers reach the 1000-2000s range.
Synthetic communities

These are ma–made and can be either simulated in silico or as sampled from an artificial mixture. They are simple, rarely go beyond a hundred species and are mainly used for understanding and testing ecological theories.
Enriched communities

Such communities stand at the intersections between simple but artificial and complex but close to naturally occuring communities. They form a sub category under artificial communities.
4. Fingerprinting
To get an idea of how unique this “key” could get we can look to how long a the typical SSH RSA key 🔑 is:
-----BEGIN PUBLIC KEY-----
MIGfMA0GCSqGSIb3DQEBAQUAA4GNADCBiQKBgQCq
GKukO1De7zhZj6+H0qtjTkVxwTCpvKe4eCZ0 FPq
ri0cb2JZfXJDgYSF6vUpwmJG8wVQZKjeGcjDOL5U
lsuusFncCzWBQ7RKNUSesmQRMSGkVb1/3j+skZ6U
tW+5u09lHNsj6tQ51s1SPrCBkedbNf0Tp0GbMJDy
R4e9T04ZZwIDAQAB
-----END PUBLIC KEY-----
This identifies your machine as you when you try to log into a host server.
Similarly a microbiome signature would look like this:
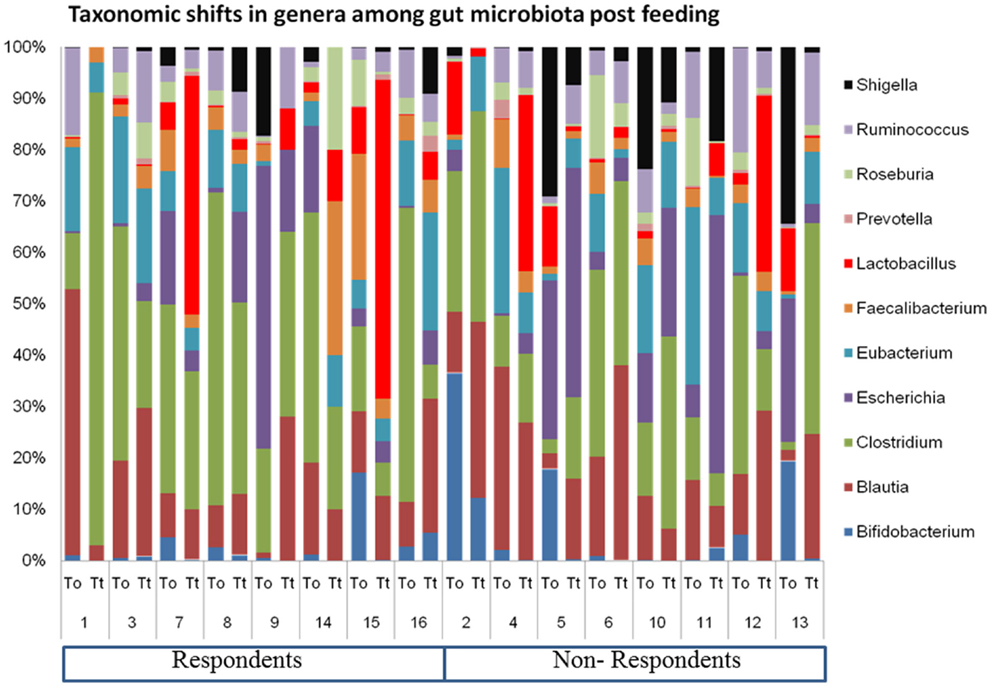
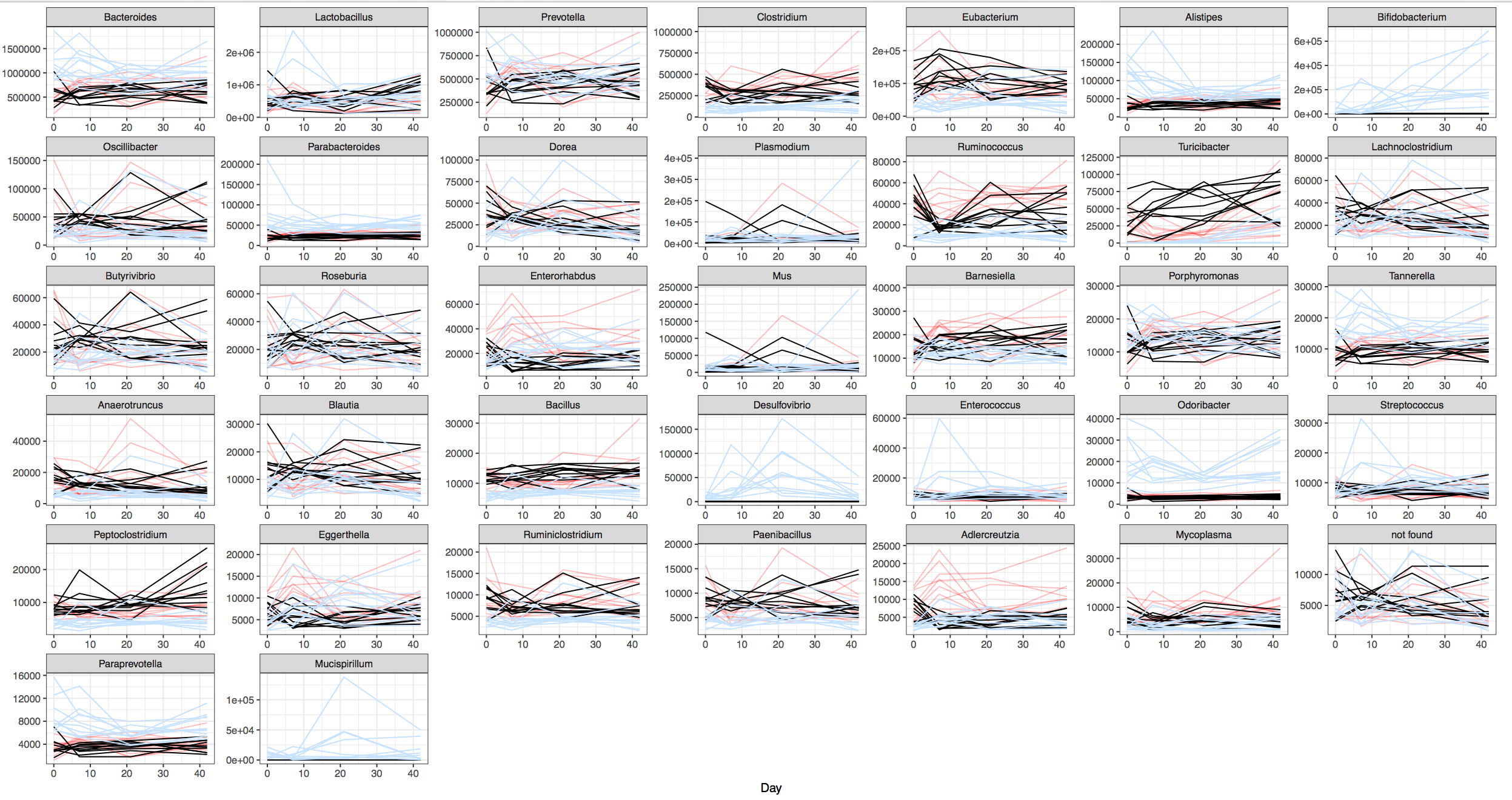
Look at how much the OTU abundances resembles a barcode:

We use this to differentiate groups of individuals from one another, usually from the diets that each individual share with others in the same group.
Take for example the plots found in the example analysis below, for the gut microbiomes from two different sets of mice each fed on a specific diet.
5 Engineer Your Gut Microbiome Now.
Ultimately, the gut microbiome is resilient towards change and will probably stay the same unless you do something drastic about your diet like going vegan as referenced in this review article. However, it is a good diagnostic to identify groups where their physiology has altered their microbiome.
Example analyses
I’m including links to a short analyses of three groups of mice, two groups fed on 2 different diets and one group which was fed a transition diet.
DISCLAIMER: The following is based on unpublished data (Little et. al). Any reproduction or use of the analysis and the results for personal/commercial use is prohibited. If you have any enquiries please contact the author of this post at wesley@bic.nus.edu.sg
Part 1: Unbiased 16s profiling of whole metagenome data using Ribotagger
Part 2: Whole Metagenome profiling
Conclusion
Things are bound to get more interestingly as studies with higher throughput time-series experiments become the norm in the near future.
References
1: Xie, C., Lui, C., Goi, W., Huson, D. H., Little, P. F. R., & Williams, R. B. H. (2016). RiboTagger : fast and unbiased 16S / 18S profiling using whole community shotgun metagenomic or metatranscriptome surveys. BMC Bioinformatics, 17(Suppl 19). http://doi.org/10.1186/s12859-016-1378-x
2: Franzosa, E. a., Hsu, T., Sirota-Madi, A., Shafquat, A., Abu-Ali, G., Morgan, X. C., & Huttenhower, C. (2015). Sequencing and beyond: integrating molecular “omics” for microbial community profiling. Nature Reviews. Microbiology, 13(6), 360–72. http://doi.org/10.1038/nrmicro3451
3: Jari Oksanen, F. Guillaume Blanchet, Michael Friendly, Roeland Kindt, Pierre Legendre, Dan McGlinn, Peter R. Minchin, R. B. O’Hara, Gavin L. Simpson, Peter Solymos, M. Henry H. Stevens, Eduard Szoecs and Helene Wagner (2016). vegan: Community Ecology Package. R package version 2.4-1. https://CRAN.R-project.org/package=vegan
comments powered by Disqus