
blast2lca++ a Python Wrapper for MEGAN blast2lca
Download now from: https://github.com/etheleon/blast2lcaPlus
“Metagenomics (also referred to as environmental and community genomics) is the genomic analysis of microorganisms by direct extraction and cloning of DNA from an assemblage of microorganisms.”
From absolute to intermediate beginners venturing into the field of Metagenomics, one tool you’ll most certainly and quickly come across is MEGAN from Daniel Huson’s Lab, Tubingen University.

If you take a closer look inside the tools directory of the installation, you’ll find a bash executable called blast2lca (see link to script on github repo)which taps into the java classes used in the desktop version of MEGAN.
blast2lca is extremely valuable as a tool for one to basically access the core algorithms within MEGAN (for example):
- Lowest Common Ancestor algorithm and
- Functional functional assignment (KEGG/COG/eggNOG)
MEGAN’s been around for awhile with its 1st release way back in (2007).
In its newest iteration MEGAN6 now includes new additions to deal with increasingly large datasets.
However, I would say the updates are still mainly for desktop users and if you need to run any huge jobs on multiple large sequencing projects, you’ll be hardpressed to find a solution unless you pay for the server edition and even then incorporating MEGAN into the customised pipeline might not be that simpl might not be that simple.
Discussions about MEGAN server will be outside of the blogpost, message the authors if you want to know more.
blast2lca++
In this blogpost, I’ll be sharing with you python wrappers https://github.com/etheleon/blast2lcaPlus, I’ve written around blast2lca. (At the time of writing I tested this with MEGAN6 community edition 6.6.0 from Dec 2016).
Use case
Say for example you’ve got a huge number of samples which you would like to run some analysis and you’re using a weak Macbook 12, but alas you have access to a powerful univeristy headless server.

One option is to install MEGAN server (Ultimate edition) run the LCA and functional binning algos there and analyses it via the desktop.
For someone who does further analyses in R and Python, that’s not really what I want to do, but do my own plots and run my own analyses.
Luckily there’s blast2lca kindly provided by the author.
However there’s still several steps which are not clear, hence the reason for this wrapper
- Combine Annotations - How does one combine KO and TAXONOMIC annotations such that we will have a combined annotation for each query (be it short read/long read/contig).
- gi2ko mapping file generator - KEGG annotations. In the Community Edition, the tools to generate the mapping file (gi kegg) is not included like in the ultimate edition. What if you’re not in a position to pay for the ultimate edition license (which bundles with it the KEGG database licence) or have a older version of KEGG lying around somewhere in the server, what should you do? (NCBI has recently done away with the GI and I’ll update this in the future)
- A complete pipeline from blast to combined output - How to go all the way from the tabbed blast output to the KO and taxonomy combined output mentioned above.
Use blast2lca++ tool of course!

Combine Annotations
In the root directory of the github repo, you’ll find a parseMEGAN python script.
It requires blast results be arranged in the following manner:
/projectDir
└── sampleDir/
└── sample.daa / sample.m8 (tabbed blast)
note if you only have one sample, then just substitute sampledir with a
..
You’ll be asked to specify the locations of the mapping files (KEGG and taxonomy as well as the path to the executable)
parseMEGAN $PROJECTDIR $SAMPLEDIR $SAMPLENAME taxOutput koOutput
After which you’ll get the outputs from blast2lca and with a merged file sample-combined.txt
/projectDir
└── sampleDirName/
├── blast2lca-tax-Output
├── blast2lca-ko-Output
├── sampleName-combined.txt
└── inputSampleDAAfile.daa
In the sample-combined.txt file, you’ll find a table:
rank ncbi-taxid KEGG-ko #reads
phylum 67820 K00000 4
phylum 1224 K06937 1
phylum 1224 K00656 6
phylum 1224 K04564 2
phylum 1224 K06934 1
phylum 1224 K12524 24
phylum 1224 K00558 7
phylum 1224 K02674 1
phylum 1224 K06694 1
phylum 1224 K01785 12
...
...
species 1262910 K00033 1
species 1262911 K00000 525
species 35760 K00000 14
species 7462 K15421 1
species 7462 K00000 1
species 1262918 K00000 365
species 1262919 K02429 5
species 1262919 K07133 1
species 1262919 K03800 1
species 1262919 K00000 529
Below’s a small example of what you could do with the data:
Application
With the above you could generate a reads per million column based on the raw counts
| level | taxon | ko | rpm | c1.raw |
| Genus | Abiotrophia | K00000 | 1.40278158 | 32 |
| Genus | Acanthamoeba | K00000 | 2.11362518 | 47 |
| Genus | Acaryochloris | K00000 | 61.73957107 | 1423 |
| Genus | Acaryochloris | K00013 | 0.00000000 | 0 |
| Genus | Acaryochloris | K00016 | 0.00000000 | 0 |
| Genus | Acaryochloris | K00091 | 0.05123577 | 1 |
You can now quickly summarise in a gene centric format the contributions made from each genus (or any taxonomic rank you choose) to each KO.
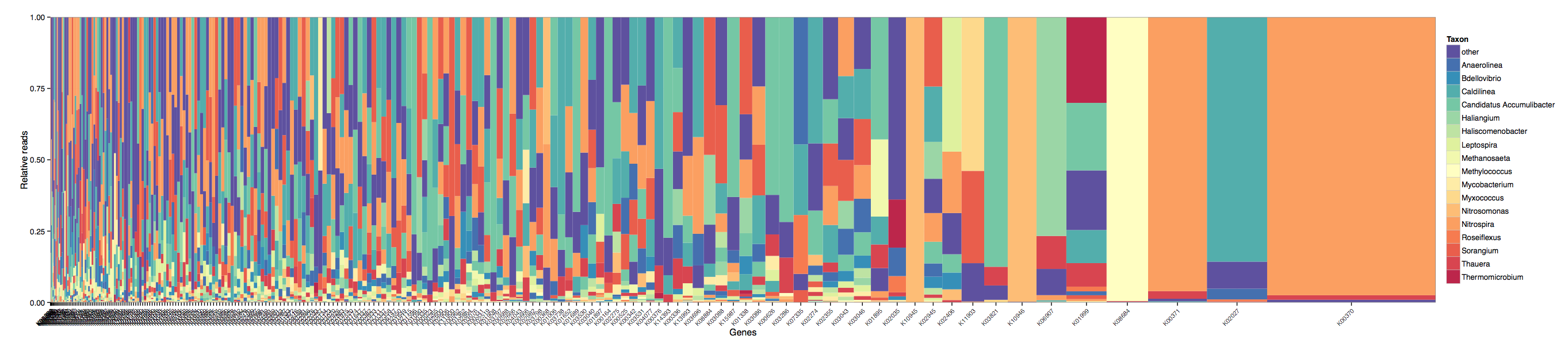
Here we see the transcriptome summary and one highest expressed KOs (right most) is a one responsible for nitrogen metabolism is mostly being expressed by a single genus (orange).
0 in the ncbi-taxid and K00000 in the KEGG-ko columns stands for unclassified.
What if you want to find out the names organisms from NCBI’s taxid?
Checkout R package MetamapsDB which lets you query the names based on taxids and do much more
Full pipeline
fullPipeline $PROJECTDIR $SAMPLEDIR $SAMPLENAME $INPUTFILE taxOutput koOutput --blast2lca <path 2 the blast2lca script> --gi2tax <path to the taxonomy mapping file> --gi2kegg <path to the KEGG mapping file>
The fullPipeline script will take a .m8 (tabbed blast) file or meganised .DAA file as input (checked via regex .m8 or .daa) and carry out the taxonomic and functional (KEGG) annotation and take the outputs and generate a combined output
gi2ko mapping file generator
Although MEGAN UE provides a KEGG mapper generating tool (not included with MEGAN CE), it doesnt take into consideration how NCBI has assigned a unique GI to each representative sequence in the non-redundant database (NCBI NR) under which are “duplicate” sequence GIs and ref IDs
when blast or diamond does the alignment it only returns the former and the rest. It makes the kegg to gi mapping irrelevent.
We’ve separately included in the tools folder of the python package the ref2kegg.go nr-gi to kegg ortholog KO mapping file generator written in golang. The output of this could be fed to the blast2lca wrapper via the --gi2kegg flag. At
the time of writing the parser is written in golang (a typed language) from perl to increase the speed of parsing the NR fasta.

Hope this helps anyone doing any customised pipeline with MEGAN6! Personally I feel strongly that MEGAN has now a open source version via MEGAN CE.
Reference
- Handelsman, J (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev., 68, 4:669-85.
- Huson, D. H., Tappu, R., Bazinet, A. L., Xie, C., Cummings, M. P., Nieselt, K., & Williams, R. (2017). Fast and simple protein-alignment-guided assembly of orthologous gene families from microbiome sequencing reads. Microbiome, 5(1), 11. http://doi.org/10.1186/s40168-017-0233-2